Elucidating the impact of apolipoprotein E and metabolic comorbidities on the pathogenesis of Alzheimer’s Disease (APOE-META-BRAIN)
This project aims to provide new insights into the roles of apolipoprotein E (apoE) in brain cell functions and to explore the relationship among apoE and metabolic comorbidities such as dydlipidemia and fatty liver on the pathogenesis and progression of Alzheimer’s Disease (AD). Understanding of this relationship may have important clinical implications in the management of patients with AD.

Alzheimer’s disease (AD) is the most common cause of dementia in the elderly (50-70% of all cases). Because of the ongoing increase in life expectancy, the prevalence of AD is expected to rise dramatically during the next 40 years in the Western world. Since little progress has been made toward finding a cure for AD, it is increasingly accepted that an in depth understanding of the cellular cascade of events leading to neurodegeneration is necessary for the design of novel therapeutic approaches for AD.
AD pathogenetic processes are related to the deposition of extracellular and intraneuronal beta-amyloid (Aβ) peptides, intraneuronal neurofibrillary tangles (NFTs) of the hyper-phosphorylated protein tau (ptau) and neuro-inflammation. The APOΕ4 isoform of apolipoprotein E (apoE), a key lipid transport protein in circulation and the brain, is the strongest and best-established risk factor for late-onset AD.
Several studies suggest that apoE4 causes AD via different processes such as disturbances of Aβ production, deposition and clearance, tau phosphorylation and aggregation, activation of neuro-inflammation, and synaptic loss. Accumulating evidence suggests that a series of metabolic comorbidities, including hyperlipidemia and non-alcoholic fatty liver disease (NAFLD) result in increased risk for AD and affect AD pathogenesis and severity. The exact role of brain apoE4 on AD initiation and progression as well as the contribution of dyslipidemia or fatty liver are not fully understood.
Our long term vision is to unravel the molecular mechanisms leading to the initiation and progression of AD. Understanding the fundamental cellular cascade of events that lead to neurodegeneration will ultimately have the highest impact on accelerating progress in disease prevention and cure.
ApoE and Alzheimer’s Disease: the role of comorbidities in AD risk
Apolipoprotein E (apoE) is glycoprotein consisting of 299 amino acids that is expressed and secreted by the liver and macrophages but is also expressed at high levels in astrocytes and microglia of the brain (1). ApoE plays a major role in lipoprotein metabolism by facilitating the clearance of lipoprotein remnants from the circulation via binding to lipoprotein receptors such as LDLr and LRP. It has three common isoforms (apoE2, apoE3, apoE4) in the general population that differ only in the amino acid positions 112 and 158 (2). ApoE4 is the strongest and best-established genetic risk factor for sporadic late-onset AD. ApoE4 has increased propensity, compared to apoE3 and apoE2, to undergo proteolytic cleavage and
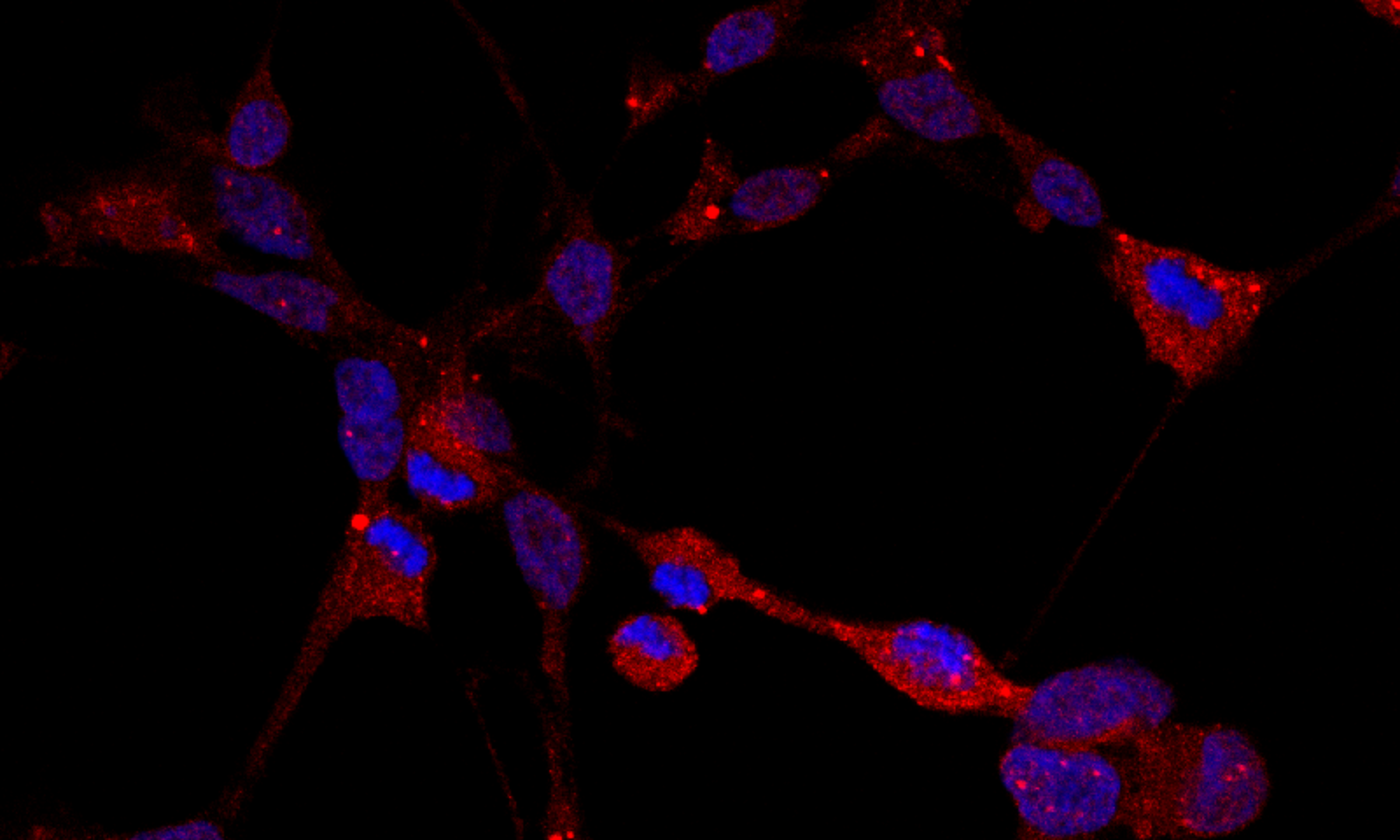
bioactive C-terminal truncated forms of apoE4 have been found in brains of AD patients and apoE4 transgenic mice (3). ApoE4 fragmentation is considered an early event in the pathogenesis of AD (4).
Our previous studies showed that specific apoE4 variants (apoE4 fragments and point mutants) are involved in events associated with AD pathogenesis, such as Aβ production and oligomerization, intraneuronal Aβ42 accumulation, Aβ phagocytosis, toxicity, oxidative stress, inflammatory responses (5-8). Collectively, findings by us and others suggest that specific apoE4 variants have different effects on AD-related pathogenetic processes and can modify distinctly the risk of disease. In addition to the established roles of secreted apoE, it has recently been shown that apoE can also be found in the nucleus and to be associated with alterations in the expression of certain genes that could have causative roles in AD (9).
AD pathogenetic processes have been linked with several comorbidities. Evidence from clinical and molecular studies suggest that chronic diseases, including hyperlipidemia, type II diabetes, non-alcoholic fatty liver Disease (NAFLD), non-alcoholic steatohepatitis (NASH) and CVD may be associated with an increased risk of AD in different populations (10). Of note we showed recently that long-term administration of sugar-containing diets, despite no effect in fasting plasma glucose or insulin levels, can enhance neuroinflammation in the AD (11). The disruption in several shared biological pathways has been proposed as the underlying mechanism for the association between AD and the aforementioned comorbidities. In addition to CVD per se, risk factors for CVD including hypertension, hyperlipidemia, and obesity have been associated with an increased risk for AD. Hepatic malfunction due to hyperlipidemia and NASH has been proposed to contribute to AD by several of non-excluding pathways, including disruption of the blood-brain barrier, failure to maintain Aβ homeostasis at the
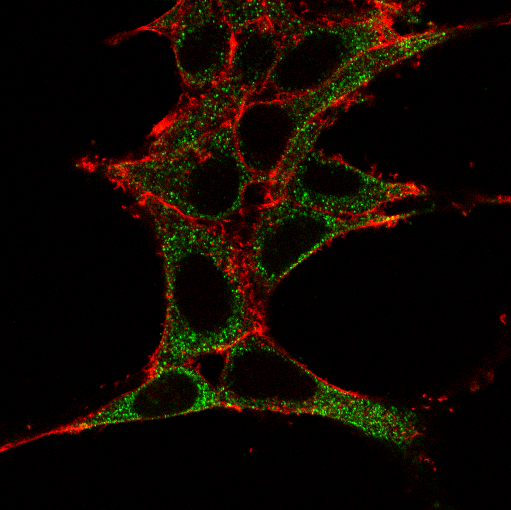
periphery; excess of pro-inflammatory cytokines due to chronic inflammation and metabolic impairment (12). ApoE, a lipid binding protein and the main lipid carrier in brain, can induce alterations of lipid homeostasis in cells, circulation and cerebrospinal fluid and could consist a link between AD and comorbidities such as hyperlipidemia, NAFLD and CVD.

References
1. Koutsodendris, N., Nelson, M. R., Rao, A., and Huang, Y. (2022) Apolipoprotein E and Alzheimer’s Disease: Findings, Hypotheses, and Potential Mechanisms. Annual review of pathology 17, 73-99
2. Serrano-Pozo, A., Das, S., and Hyman, B. T. (2021) APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. The Lancet. Neurology 20, 68-80
3. Munoz, S. S., Garner, B., and Ooi, L. (2019) Understanding the Role of ApoE Fragments in Alzheimer’s Disease. Neurochemical research 44, 1297-1305
4. Brecht, W. J., Harris, F. M., Chang, S., Tesseur, I., Yu, G. Q., Xu, Q., Dee, F. J., Wyss-Coray, T., et al. (2004) Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J.Neurosci. 24, 2527-2534
5. Dafnis, I., Argyri, L., Sagnou, M., Tzinia, A., Tsilibary, E. C., Stratikos, E., and Chroni, A. (2016) The ability of apolipoprotein E fragments to promote intraneuronal accumulation of amyloid beta peptide 42 is both isoform and size-specific. Sci. Rep. 6, 30654
6. Dafnis, I., Raftopoulou, C., Mountaki, C., Megalou, E., Zannis, V. I., and Chroni, A. (2018) ApoE isoforms and carboxyl-terminal-truncated apoE4 forms affect neuronal BACE1 levels and Abeta production independently of their cholesterol efflux capacity. Biochem. J. 475, 1839-1859
7. Dafnis, I., Argyri, L., and Chroni, A. (2018) Amyloid-peptide beta 42 Enhances the Oligomerization and Neurotoxicity of apoE4: The C-terminal Residues Leu279, Lys282 and Gln284 Modulate the Structural and Functional Properties of apoE4. Neuroscience 394, 144-155
8. Mountaki, C., Dafnis, I., Panagopoulou, E. A., Vasilakopoulou, P. B., Karvelas, M., Chiou, A., Karathanos, V. T., and Chroni, A. (2021) Mechanistic insight into the capacity of natural polar phenolic compounds to abolish Alzheimer’s disease-associated pathogenic effects of apoE4 forms. Free radical biology & medicine 171, 284-301
9. Theendakara, V., Patent, A., Peters Libeu, C. A., Philpot, B., Flores, S., Descamps, O., Poksay, K. S., Zhang, Q., et al. (2013) Neuroprotective Sirtuin ratio reversed by ApoE4. Proc Natl Acad Sci U S A 110, 18303-18308
10. Santiago, J. A., and Potashkin, J. A. (2021) The Impact of Disease Comorbidities in Alzheimer’s Disease. Frontiers in aging neuroscience 13, 631770
11. Dafnis, I., Mountaki, C., Fanarioti, E., Mastellos, D. C., Karvelas, M., Karathanos, V. T., Tzinia, A., Dermon, C. R., et al. (2022) Temporal Pattern of Neuroinflammation Associated with a Low Glycemic Index Diet in the 5xFAD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 59, 7303-7322
12. Estrada, L. D., Ahumada, P., Cabrera, D., and Arab, J. P. (2019) Liver Dysfunction as a Novel Player in Alzheimer’s Progression: Looking Outside the Brain. Frontiers in aging neuroscience 11, 174
The research project is implemented in the framework of H.F.R.I call “Basic research Financing (Horizontal support of all Sciences)” under the National Recovery and Resilience Plan “Greece 2.0” funded by the European Union – NextGenerationEU (H.F.R.I. Project Number: 15529).